Guidelines for Referring Providers
Prior Mild Contrast Reaction – Premedication Protocol
| Adult (or Pediatric > 50 kg) | Pediatric ( < 50 kg) |
|---|---|
OR
|
OR
|
Prior Moderate, Severe, or Unknown Severity Contrast Reaction – Premedication Protocol
| Adult (or Pediatric > 50 kg) | Pediatric ( < 50 kg) |
|---|---|
|
Premedication with corticosteroid and antihistamine
AND
|
Premedication with corticosteroid and antihistamine
AND
|
IV Alternatives for Patients Who CANNOT Take Oral Medications
| Adult (or Pediatric > 50 kg) | Pediatric ( < 50 kg) |
|---|---|
|
|
Accelerated Premedication Protocol
For use in patients with prior moderate, severe, or unknown contrast reaction when clinical situation warrants imaging sooner than allowed (12 hours) with standard protocol.
| Adult (or Pediatric > 50 kg) | Pediatric ( < 50 kg) |
|---|---|
AND
|
AND
|
Extravasation
Patients with extravasation should be assessed and reassured by a radiologist, and referred to the Emergency Department, if there is skin blistering, altered tissue perfusion, increasing pain, or change in sensation distal to the site of extravasation. In all cases, it is critical that the responsible radiologist communicates directly with the patient, referring physician, and Emergency Department as appropriate and documents these communications in the report or medical record.
For more information about Contrast Premedication, visit:
Iodinated CT: Guidelines for IV Iodinated Contrast Administration for CT Exams
| Est. GFR ml/min/1.73 m2 |
Guidelines for Contrast Administration and Hydration |
|---|---|
| ≥ 30 | Low risk. At the current time, there is very little evidence that intravenous iodinated contrast material is an independent risk factor for AKI in patients with eGFR ≥ 30 mL / min/1.73m2. |
| < 30 |
Higher risk. This cohort of patients appears to be at greatest risk for post-contrast acute kidney injury after administration of intravenous iodinated contrast. Contrast should not be administered unless the patient is on dialysis and anuric, or if contrast is considered diagnostically imperative and the benefits of contrast outweigh the risk of post-contrast acute kidney injury. If the patient meets these criteria, the referring attending physician should document the need for contrast and that the benefit of contrast outweighs the risk of post-contrast acute kidney injury in the patient’s medical record. Pre-procedural prophylaxis again post-contrast acute kidney injury with intravenous volume expansion therapy should be utilized. The optimal IV volume expansion protocol is unknown and ideally should be tailored to the patient’s volume status and medical conditions, which may necessitate discussion between the referring physician and the radiology team. Suggested protocols:
|
For more information about Iodinated CT, visit:
Guidelines for IV Iodinated Contrast Administration for CT Exams
Patients on dialysis can receive IV contrast, but the fact that a patient is on dialysis should NOT be regarded as automatically allowing the administration IV contrast, because of several potential hazards, including:
- In the setting of acute renal failure, where dialysis is being performed with the expectation of renal recovery, it may be inappropriate to administer a nephrotoxic agent that may jeopardize the reversal of renal impairment.
- In the setting of chronic renal failure where patients are still producing a small amount of urine, the small amount of residual renal function could be imperiled by IV contrast, potentially increasing the required frequency of dialysis and hastening the complications of severe renal impairment – neither of which are trivial considerations. Patients with renal insufficiency who require only intermittent or occasional dialysis are at substantial risk for contrast media-induced nephrotoxicity with further worsening of their renal function. Alternative imaging studies not requiring contrast media should be strongly considered.
- In either setting, the volume of IV contrast may add to fluid overload, potentially adding to circulatory compromise. The volumes of both oral and IV contrast should be included in the fluid intake of dialysis patients.
While these hazards of giving IV contrast to dialysis patients may be relatively small, these risks should be weighed against the likely diagnostic benefit of contrast administration. The Nephrology Service is readily available for consultation in cases where the risk/benefit assessment is complicated, and closely follows all hospitalized dialysis patients.
It should also be noted that the common belief that dialysis patients require early post-procedural dialysis is unsupported by clinical studies and expert guidelines. Dialysis pre-procedure may be desirable, particularly if a large dose of contrast is anticipated or in patients with heart failure.
Metformin (Glucophage®) is an oral hypoglycemic agent that is eliminated by renal excretion. The most significant potential adverse effect of metformin therapy is the development of metformin-associated lactic acidosis, a rare but serious condition that can be fatal. Any factors that decrease metformin excretion from the kidney or increase blood lactate levels may theoretically increase the risks for patients to develop metformin-associated lactic acidosis.
Prior guidelines from the American College of Radiology recommended withholding metformin when a patient was planned to receive intravascular iodinated contrast based on the theoretical risk of patients developing contrast-induced nephropathy and therefore retaining metformin within the body. However further examinations of the rare cases of metformin-associated lactic acidosis have revealed that almost all cases occurred when patients were receiving metformin despite having one or more patient-associated contraindications to receiving this drug. There have been no reports of lactic acidosis in patients properly selected for metformin therapy. As a result, recent guidelines from the American College of Radiology are more measured in the management of this medication around the time of intravascular iodinated contrast administration:
Category I
In patients with no evidence of AKI and with eGFR ≥30 mL / min/1.73m2 , there is no need to discontinue metformin either prior to or following the intravenous administration of iodinated contrast media, nor is there an obligatory need to reassess the patient’s renal function following the test or procedure.
Category II
In patients taking metformin who are known to have acute kidney injury or severe chronic kidney disease (stage IV or stage V; i.e., eGFR< 30), or are undergoing arterial catheter studies that might result in emboli (atheromatous or other) to the renal arteries, metformin should be temporarily discontinued at the time of or prior to the procedure, and withheld for 48 hours subsequent to the procedure and reinstituted only after renal function has been re-evaluated and found to be normal.
Reference
- ACR Manual on Contrast Media Version 10.3, 2017: 47-49. American College of Radiology.
For more information about Contrast Administration, visit: (anchor link to metformin section)
Guidelines on the Administration of Intravenous Gadolinium-Containing Contrast Media (UCSF Department of Radiology Gadolinium Policy)
Overview
- Gadolinium-based contrast agents (GBCAs) should only be administered when deemed necessary by the radiologist.
- Routine screening and laboratory testing for renal failure is no longer required prior to the administration of group II agents.
- If a patient presents with known renal failure, the necessity of a group II agent should be confirmed by the radiologist.
- If a group II agent is used in the setting of dialysis, hemodialysis should be performed as soon as possible after contrast administration.
- Group I agents are contraindicated in patients on dialysis, and are no longer used at UCSF.
- Group III agents (Eovist®) require informed consent when eGFR < 30
|
|
eGFR > 30 |
eGFR < 30 |
|
Group II GBCA |
Single dose appropriate |
Confirm necessity of GBCA |
|
Group III GBCA |
Single dose appropriate |
Informed consent needed |
For more information about MRI with Contrast (Gadolinium-Containing) Policy, visit:
The risk of NSF is extremely low when group II agents are used in the setting of dialysis.1 Dialysis after GBCA administration, however, does not protect patients from developing NSF.2 Studies have shown that the serum concentration of gadolinium is significantly decreased after hemodialysis, but there is no information regarding residual tissue amounts.2 Theoretically, the sooner the dialysis session is performed the less amount of contrast agent is deposited in the tissues. Therefore, all patients already receiving dialysis treatment should be scheduled for hemodialysis as soon as practical following the gadolinium-enhanced MRI and preferably within 24 hours. Patients receiving peritoneal dialysis do not need to be switched to hemodialysis.
This should be arranged by the requesting physician in consultation with the patient’s outpatient nephrologist and dialysis unit. Routine MRI studies should be scheduled in the morning and dialysis scheduled in the afternoon following the study; radiology scheduling staff will give morning slot priority to dialysis patients. Administration of dialysis promptly after gadolinium may require altering the patient’s regular outpatient dialysis schedule and advance communication several days in advance with the nephrologist and dialysis unit. There is general consensus that a patient with chronic kidney disease who is not already dialysis dependent should not be started on dialysis after administration of gadolinium for precautionary purposes only, since there is no data to support the benefits of this intervention.
Key point: Dialysis should preferably be performed within 24 hours of gadolinium administration to patients already on dialysis. The institution of dialysis is not required in patients with severe renal impairment who are not already on dialysis after gadolinium administration.
For more information about creatinine testing prior to gadolinium administration, visit:
Implantable Pediatric Sternum Device
A new implanted sternal device system for pediatric patients is contraindicated for MRI. Dr. Michael Harrison (pediatric surgery UCSF) is currently running a phase 2 research procedure, The Magnetic Mini-Mover procedure. This research procedure utilizes the following products, “Magnimplant” and “Magnatract” in a combined system to correct for pectus excavatum or sunken chest deformity. An internal magnet is surgically placed behind the sternum for "Magnimplant," and “Magnatract” is an external brace with a magnet implanted. The devices combine to exert a magnetic force on the chest wall.
This device system is totally contraindicated for MRI at any field strength. Per Dr. Harrison: “The device is made for us by Texcel and stays in 18-30 months.” “It is implanted on the sternum through a small subxiphoid incision which will always be visible, so this would be the best marker in an unconscious patient.” “The patients are teen and preteen and they and their parents have verbal and written warnings about MRI. In addition the patients wear (or at least are given) a medi alert bracelet saying no MRI.” Parents of pediatric patients should specifically be asked if this magnet device has been implanted. Most devices have been for older children, but they may be seen in patients as young as 1 year old in the future. The devices may remain implanted for years, although studies are still ongoing to determine the best clinical usage. More information is available on the Pediatric Surgery website and the National Library of Medicine. Contact the principle investigator Dr. Michael Harrison with further questions.
Note that MRI can be performed if a patient has a Nuss bar for pectus excavatum repair made of titanium alloy plates.
Metallic Foreign Body in the Eye
Intra-ocular metal foreign bodies are a cause of major concern in MR safety. It is not uncommon for patients who have worked with sheet metal to have metal fragments or slivers located in and around the eye. Since the magnetic field exerts a force on ferromagnetic objects, a metal fragment in the eye could move or be displaced and cause injury to eye or surrounding tissue. A screening CT of the orbits for foreign body must be cleared by a radiologist prior to the patient entering the scan room (Zone 4).
"Triggerfish" Contact Lens
A new contact lens that allows for an automated recording of continuous intraocular pressures has come to market SENSIMED Triggerfish®. This device is MRI unsafe, and can result in severe eye burn.
Gastric Reflux Device
The LINX Reflux Management System is a series of titanium beads with a magnetic core implanted around the lower end of the esophagus to control gastro esophageal reflux disease (GERD). This implant is totally contraindicated for MRI in both the 1.5T and 3T. The FDA website has more information.
Insulin Pumps
There are two basic types of insulin pumps, one is used as an external device and the other is implanted. Both types currently pose hazards to patients referred to MRI procedures. For an external insulin pump, in general, the device typically needs to be removed and kept out of the MRI environment to ensure that there is no adverse impact on the functionality of the external pump. The implanted pump will be adversely affected by the magnetic field and will need to be removed prior to imaging.
Temporary Transvenous Pacing Leads
Temporary external transvenous pacing leads contain nonferromagnetic, but electrically conductive material. Radiofrequency pulses in MRI can induce currents that could lead to thermal injuries. They are contraindicated for MRI. The same is true for abandoned intracardiac pacing leads.
General
Shrapnel
Most pellets and bullets tested for MR compatibility are composed of nonferromagnetic materials. Ammunition that is ferromagnetic tended to be manufactured in foreign countries and or used for military applications. Because pellets, bullets, and shrapnel may be contaminated with ferromagnetic materials, the risk versus benefit of performing an MR procedure in a patient should be carefully considered, as well as whether or not the metallic object is located near a vital anatomic structure. Shrapnel within or adjacent to the globe in the orbit is an absolute contraindication. However, outside of this area most shrapnel is embedded within scar and unlikely to experience significant torque when placed in a magnetic field. Radiographic screening is recommended in patients with a history of gunshot wounds. Plain film radiographs are usually sufficient to confirm that metallic debris is located within subcutaneous, intraosseous or otherwise non-vital tissues; however, suspected proximity to large arteries or viscera should be further investigated with low-dose noncontrast CT when there is ambiguity on plain films.
Pregnancy
For more information, visit:
Implantable Drug Infusion Pumps
Drug infusion pumps are used for automatic delivery of antineoplastic agents or narcotics. While some drug delivery devices are "constant flow" without a motor, infusion pumps have a motor that may have ferromagnetic properties, a magnetic switch, and are programmable by telemetry. Potential MRI issues include:
- Opening of outlet valves resulting in discharge of the entire drug reservoir.
- Temporary or permanent stall of the motor.
- Local tissue heating and increased potential for peripheral nerve stimulation (PNS).
- A policy is being developed with the Pain Management Service to expedite the scanning of patients with Medtronic Synchromed II pumps, which are MRI conditional at 1.5T. The identity of the pump must be documented before the MRI scan, and then confirmed at the time of the scan.The pump program needs to be check after the MRI: for outpatients, this can be done at a pre-scheduled appointment with the managing service; inpatients will need to have the Pain Management team perform this check.
- Constant flow systems (Codman, Medtronic Isomed) are MRI conditional at 1.5T.
- Medallion pumps are investigational at this point, but are reported to be MRI conditional at 1.5 and 3T.
- Codman MedStream pumps are MRI conditional at 1.5 and 3T. Pump program needs to be checked after MRI.
- Flowonix Prometra pumps should never be scanned at UCSF.
If there is any doubt about the suitability of a device for MRI, or about management of a device before and after MRI, contact the Neuromodulation Service (lawrence.poree@ucsf.edu). Below are x-ray images of the common Synchromed II pump and the two pumps that should not be imaged without outside consultation and management: Prometra I and II pumps.

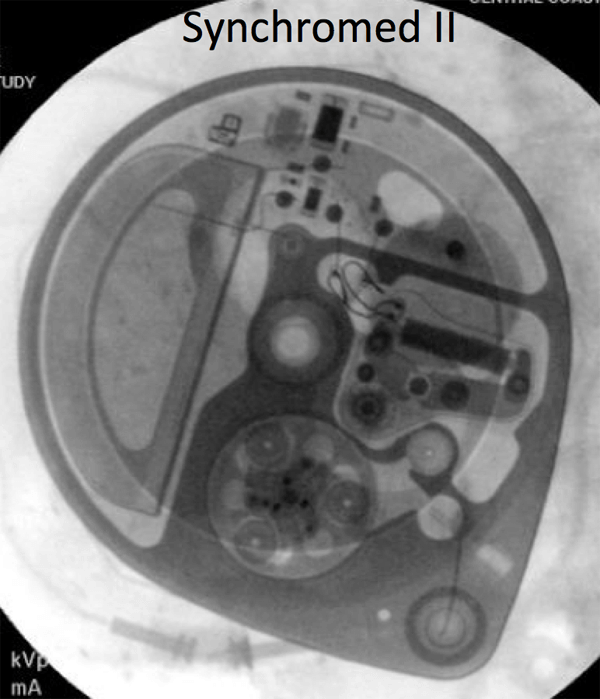
Epidural Catheters
Many epidural and peripheral nerve catheters contain conducting wire that could heat during magnetic resonance imaging (MRI). The Arrow® FlexTip Plus® Epidural catheter is currently being used by neurosurgery and anesthesia here at UCSF and this catheter is rated as MRI conditional with the following imaging limitations:
- Static magnetic field of 3T or less.
- Maximum spatial gradient magnetic field of 720-Gauss/cm or less.
- Use of a transmit/receive head RF coil only.
MRI with this catheter in position is thus safe only for examinations of the head. All other epidural catheters must be researched with the manufacturers for contraindication or conditional imaging guidelines. If there is no information available pertaining to the epidural catheter, it must be removed prior to imaging.
Feeding Tubes
One of the feeding tubes (Mclean-Ring Enteral Feening Tube) used at UCSF is not safe for MRI.
The commonly used Corflo feeding tube (the "yellow" tube, including those with tungsten-weighted tips) is MRI safe at 1.5T and 3T with the stylet removed.
Spinal Fixation Hardware
The main issue with imaging spinal fixation hardware (including Harrington rods) is artifact. Heating is also possible. Imaging at 1.5T is preferred with careful attention to patient padding in order to prevent skin-to-skin contact points. Patients should be conscious during imaging to report any unusual heat sensation. If only 3T imaging is available, normal operating mode (SAR of 2 W/kg) should be used.
Neuro
Halo
Halo vests pose several risk factors which include deflection and subsequent dislodging of the halo, heating due to RF absorption, electrical current induction within the halo rings, electrical arcing and severe art factual consequences which could render the imaging acquisition useless. Non-ferrous and non-conductive halo vests, which are MR compatible, are commercially available.
Neuro-Stimulation Systems
There are two general types of neurostimulators:
- Passive receivers: neurostimulators that receive RF energy that is magnetically coupled from an external device by means of a coil placed over the implanted device.
- Hermetically encased pulsed generators: neuro-stimulators that contain a battery and are programmed by an external device to produce the various stimulus parameters.
Exposure of a neurostimulator to the electromagnetic fields used for MR imaging may produce device malfunction and or harm to the patient. Neuro-stimulation systems include the following: Deep Brain Stimulator (DBS), Vagus Nerve Stimulator (VNS), and Spinal Cord Stimulator (SCS). Each device has manufacturer’s conditional imaging guidelines regarding: field strength and RF coil restrictions; SAR limitations; and scan area restrictions.
MRI with these devices should only be performed in strict accordance to manufacturer’s conditional guidelines. If there are abnormal impedances, neurology should add language to acknowledge the impedance issue and their discussion about this with the patient in their notes. A policy is being developed with the UCSF Neurology Service that would allow access to MRI for patients with Medtronic DBS Neurostimulators, which can either be "head-only" or "full-body" eligible:
- Head-only eligible: Model 37602 (Activa SC), Model 7428 (Kinetra), Model 7426 (Soletra).
- Full-body eligible (after 2009): Model 37612 (Activa RC), Model 37603 (Activa SC), Model 37601 (Activa PC).
Scanning of these devices can be performed on the Philips 1.5T (MR4), or on 3T systems that allow monitoring of B1 rms (although not after a complete device has been placed), using the following protocol:
- When an MRI study on a patient with a deep brain stimulator (DBS) is requested, it will be flagged by scheduling and placed in the MRI Manager’s work queue.
- An MR Manager will assess whether a Medtronic DBS Eligibility form has been completed.
- If the elegibility form has been completed, and the device is appropriate, MRI will be scheduled on the Philips 1.5T or an appropriate 3T scanner (go to step 5).
- If there is no completed screening form, an appointment with Monica Volz in Neurology is required (415-353-7382; monica.volz@ucsf.edu). If the study is scheduled within one week of the Neurology appointment, then skip to step 6.
- Once the MR exam has been scheduled, the Medtronic team will be notified (donna.gow@medtronic.com; 415-370-3555) and asked to appear during the patients MR screening to assess DBS function.
- The MRI scanning protocol should be defined by the attending radiologist. If a conforming protocol does not exist, consult with Alastair Martin (415-353-8770), who will help adapt the protocol to comply with Medtronic limits (for full body eligible devices: < 2.0uT B1+rms, maximum active scanning time of 30 minutes within a 90 minute window).
- On the day of the MRI, the Medtronic DBS screening form must either exist in Apex (uploaded by Neurology) or be presented in hardcopy by the patient. The MR tech will review the screening form and make a note in Apex that it was reviewed and everything is in order. If a hardcopy form was presented it will be scanned and uploaded into Apex.
Bone Fusion (Spinal) Stimulator
These devices usually have an external electronic component that attaches to electrodes implanted in areas of fractured bones. They are used to enhance and facilitate the rate of bone healing. Review each manufacturer’s guidelines for MRI contraindications or conditional restrictions. More information may be found at mrisafety.com
Cochlear Implants
MR procedures are strictly contraindicated in patients with certain cochlear implants because of the possibility of injuring the patient and/or permanently damaging or altering the function of the cochlear implant devices and/or electrode arrays. However, there have recently been several implants that have received FDA conditional status for MRI. Patients with these cochlear implants can undergo MRI if the following criteria are observed:
- Only approved Baha and MedEL Cochlear Implants (i.e., Sonata, Pulsar, Concert).
- Only at 1.5T.
- Pre-MRI CT of the temporal bone is required for bone thickness.
Intra-Cranial Vascular Clips
Some intracranial aneurysm clips are absolute contraindications to MRI. The surgical management of intracranial aneurysms and arteriovenous malformations by the application of aneurysm clips is a well-established procedure. The presence of an aneurysm clip in a patient referred for a MR procedure represents a situation that requires the utmost consideration because of the associated risks.
Certain types of intracranial aneurysm clips (e.g., those made from martensitic stainless steels such as 17-7PH or 405 stainless steel) are prone to torque in a MR produced magnetic field. The displacement of these clips may damage the vessel, resulting in hemorrhage and or death. Intracranial clips made of titanium- or chromium-based alloys are now commonly used and have proved safe for MR.
To minimize the possibility of inadvertently imaging a patient with a magnetically active metallic implant, implanting physicians must provide patients with information about the type and identity of their particular implants, and suggest that the patients carry an alert card, or wear a medical alert bracelet,or necklace, identifying them as having such an implant. In addition, physicians who order MR procedures must carefully screen patients and inform the MR imaging facility of any metallic implants the patient may have and provide written documentation stating the name and model number of the clips.
EEG Electrodes
Traditional EEG electrodes are ferromagnetic and contraindicated for MRI. MRI conditional electrodes have recently been developed and being considered for use at UCSF. EEG electrodes that are MRI conditional afford several advantages:
- Skin breakdown and risk of infection are minimized, as electrodes are not disconnected and then reapplied.
- Patient care delays are minimized because there is no need to wait until after an MRI to apply the electrodes.
- MRI staff does not need to wait for an EEG tech to remove the electrodes.
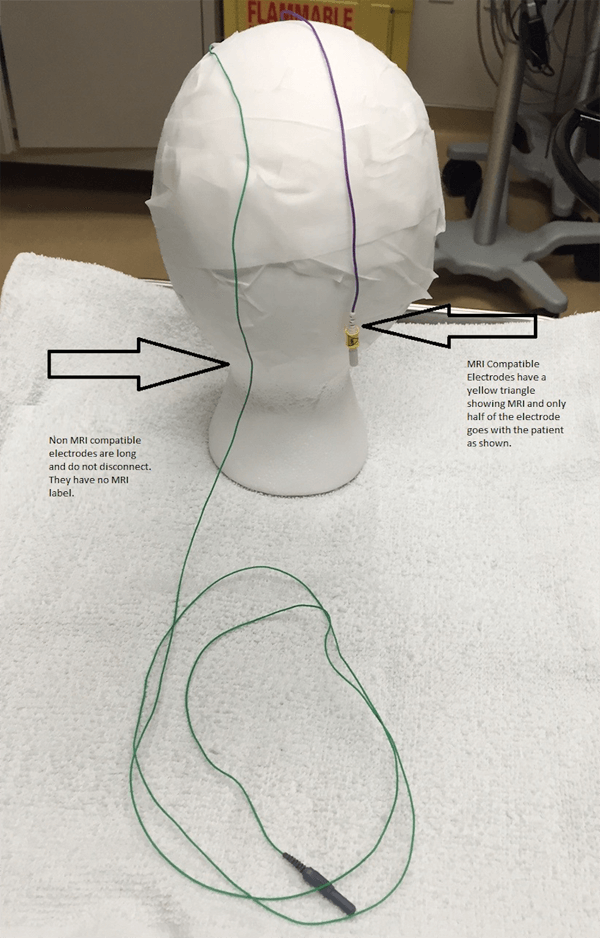
Examples of the two types of MRI conditional electrodes are pictured below. They disconnect/reconnect quickly, and only the MRI safe portion goes with the patient. An EEG tech will check each patient to make sure only MRI safe leads are going with the patient. An orange coban or a sticker marked with “MRI OK” will be applied to confirm that this check has been completed.
Non-clinical testing of these FDA cleared electrodes has demonstrated that these electrodes can safely remain on a patient during an MRI that contains multiple sequences, as long as a sequence does not exceeed 15 minutes of scan time, and are approved for both 1.5T and 3T. Other stipulations for use are:
- Maximum spatial gradient field of 4,000 gauss/cm (40T/m).
- Maximum whole body SAR of 2W/kg.
- Extension cables much be removed before entering the MRI, as they are unsafe.
Image artifact from the EEG electrodes typical extends no more than 3 mm from the skin attachment at 3T with a gradient echo pulse sequence. More details and safety data can be found at the manufacturers’ sites: Ives EEG Solutions or Rhythmlink.
For questions regarding a particular patient with EEG electrodes, contact Dawn Deines at Mission Bay (415) 361-8242 or Tom Thompson at Parnassus (415) 203-4453.

Ventricular Catheters
The Camino Flex Ventricular Catheter is used at UCSF and is MRI safe if imaged in the following way:
- Confirm the identity of the catheter.
- 1.5T or 3T is OK.
- Use only the following coils: Transmit / Receive Body Coils; Transmit Body Coils / Body Receive-Coil; Transmit Body Coils / Head Receive Coil.
- Maximum SAR of 2W/kg, or 4W/kg if using first level controlled operating mode.
- Maximum spatial gradient magnetic field of 3500G/cm (35T/m).
- Catheter should have a "coiled" configuration. If this configuration is lost, the MRI scan should be aborted.
Cardiac/Body
Breast Tissue Expanders
Tissue expansion with an inflatable breast implant is a common breast reconstruction technique. These devices typically have an injection site that may be metallic, and even magnetic in some cases. Possible complications with MRI include:
- Artifact that may prevent imaging of adjacent breast tissue.
- Movement that may cause patient discomfort and possibly displacement of the device.
- Burning sensation.
The presence of breast tissue expanders, date of placement and the device manufacturer should be documented in all patients prior to MRI. A chest x-ray may be useful to determine if the device has a metallic component. Imaging at 1.5T may be appropriate in some circumstances if the benefits of imaging clearly outweigh the risks, and the patient is aware of the possible complications and consents to proceed. The following breast tissue expanders are considered MRI unsafe:
- MAGNA-SITE
- Contour Profile Tissue Expander
Prosthetic Heart Valves
MRI is not considered hazardous for patients with prosthetic heart valves or annuloplasty rings. The attractive forces exerted on the prostheses are small compared to the force exerted by the contracting heart. There have been no reports of injury from MRI performed in patients with heart valve prostheses or annuloplasty rings.
Pacemakers, ICDs, Pacing Wires and Loop Recorders
For more information, visit:
Penile Implants
Some penile implants exhibit ferromagnetic qualities and are subject to substantial deflection forces measured when exposed to a 1.5 Tesla magnetic field. Although unlikely to cause significant injury, MRI in patients with these implants would undoubtedly be uncomfortable. For this reason, subjecting a patient with one of these implants to an MR procedure is inadvisable. There are a number of MRI conditional penile implants safe at both 3.0T and at 1.5T. Manufacturer guidelines should be followed for these devices.
Foley Catheter with Temperature Probe
A Foley catheter with a temperature probe has a thermistor or thermocouple located on or near the tip of the device and a wire that runs the length of the catheter to a connector that plugs into a temperature monitor. Sometimes an additional external cable is also used, which may or may not be removable. A Foley catheter with a temperature sensor should never be connected to an external cable and/or the temperature monitor during imaging.
Due to the possibility of heating of the wire in the Foley and possible harm to the patient, specific labeling instructions for a given Foley catheter with temperature sensor must be carefully followed. Importantly, not all Foley catheters with temperature sensors are acceptable for patients undergoing MR procedures. The catheters must be FDA approved for use in the MRI scanner. Otherwise, the temperature probe and the corresponding wire must be removed prior to imaging.
The Bardex Latex-Free Temperature-Sensing 400-Series Foley Catheter is commonly used at UCSF and is MRI conditional. For imaging, please:
- Confirm identity of the Foley Catheter.
- Position catheter with temperature sensor in a straight configuration down the center of the MRI table (i.e., aligned with the "z" axis).
- Any removable catheter connector cable should be disconnected.
- 1.5T or 3T is OK with the following stipulations: SAR of 3.5 W/kg (or 3.0 W/kg at 3T) for 15 min of scanning per sequence; spatial gradient magnetic field of 720-Gauss/cm or less.
Two other Foley catheters with temperature probes have been FDA approved for MRI, and may also be used at UCSF:
- Bard Lubricath Temperature-Sensing Foley Catheter, Ref 909116M.
- DeRoyal Foley Catheter with Temperature Sensor.
- Date of coronary stent placement and device manufacturer should be documented prior to MRI.
- If confirmed coronary stent details remain unknown despite reasonable attempts to locate them, proceed with MRI at either 1.5T or 3T.
- All current, commercially available coronary stents may be imaged at 1.5T or 3T at any time:
- Maximum whole-body-averaged specific absorption rate (SAR) of 2-W/kg in Normal Operating Mode.
- Maximum 15 min of scanning (per sequence).
As there are no known coronary stents made from ferromagnetic metallic materials, it is not necessary to wait 6 weeks or longer for MRI scanning.
Cardiac Electronic Implanted Device (CEID), cardiac pacemaker/implantable cardioverter defibrillator (ICD) in place for more than 6 weeks is strongly preferred. 1.5T scanners are the default option. 3T scanners will only be considered for appropriate MRI conditional devices. Please review cardiac pacemakers/ICD workflow diagram below for overview of how these cases should be handled. Studies will be performed only on MRI conditional devices at San Francisco Veterans Affairs Health Care System. Note that scheduling of outpatients should occur after the "vetting" stage.
6 weeks waiting period facts
- There are some manufacturers that do not require a 6 week waiting period. Referencing current manufacturer's website is recommended.
- There are limited studies showing successful MRI completion in patients without a waiting period, assessment by a MD is required, along with careful attention to limiting patient body/arm movement during positioning for the MRI.
- Pacing leads are electrically conductive, radiofrequency pulses experienced in MRI imaging can induce currents that may result in thermal injuries and severe patient harm.
- Lead heating can be very difficult to predict, as it depends on many factors:
- configuration/orientation
- polarity
- position in the patient AND in the transmit coil
- composition/structure
- shielding/sheathing
- length (resonant heating), resulting in arrythmias
- size and position of patient within the magnet bore
CEID, pacemaker/ICD lead scenarios with guidance
- Temporary epicardial pacing wires - IF cut at the skin, may undergo MRI of any body part at 1.5T or 3T.
- To clarify: these are used temporary epicardial pacing in the setting of cardiac surgery. If the leads cannot be extracted at the end of the procedure, they are left in place and cut at the skin.
- Temporary transvenous pacing leads - are an absolute contraindication to MRI due to increased heating risk (20°C/68°F) and increased risk of current induction.
- Fractured Leads - may be considered according to the non-conditional CEID protocol @ 1.5T, radiologist assessment for indication, risk vs benefit discussion with patient and signed consent.
- Abandoned, in situ, permanent intracardiac pacing leads - may be considered MR Conditional @ 1.5T, using the Non-MRI-Conditional CEID. Radiologist assessment for indication, risk vs benefit discussion with patient and signed consent.
- Abandoned, in situ, epicardial leads - seen infrequently in adults but are common in pediatrics are treated the same as intracardiac leads that are abandoned
- Image artifacts - are a risk when the lead is within imaging area.
- CEID images with lead types:
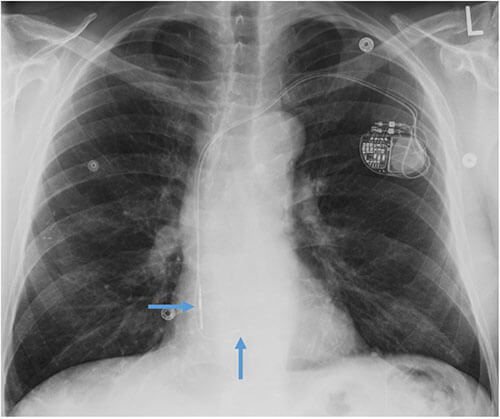

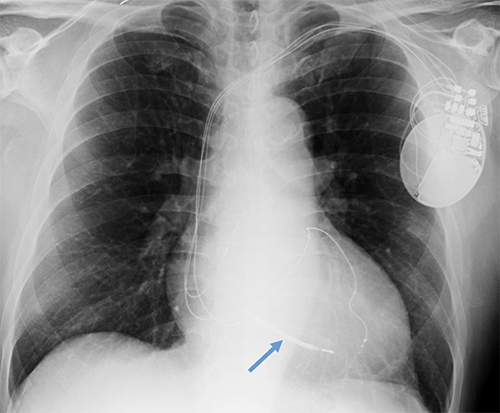
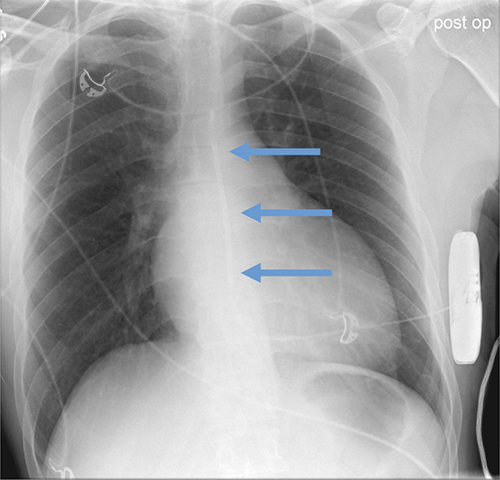
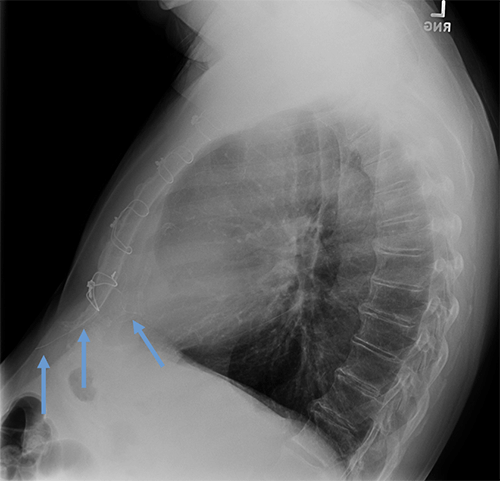
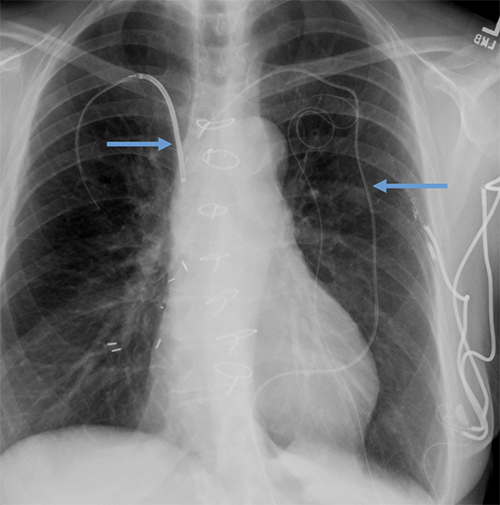
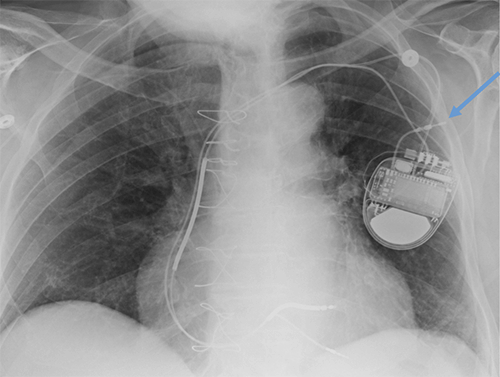
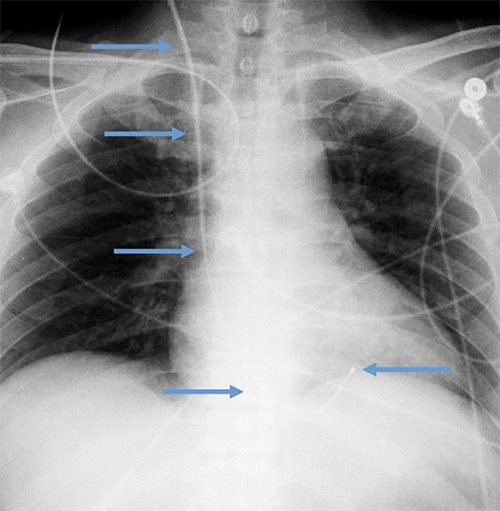
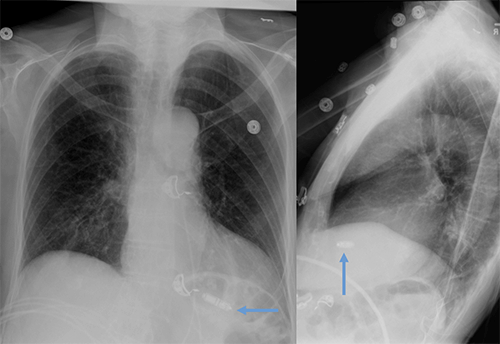
Abandoned, in situ intracardiac and epicardial leads.
At UCSF, we perform Cardiovascular Magnetic Resonance Imaging in patients with abandoned pacing leads according to our institutional non-MRI-conditional protocol following the same safety protocols as patients with leads attached to generators. This includes static field strength of 1.5T and normal operating mode - ex: SAR (≤1.5 or ≤ 2 W/kg) and dB/dt limitations (<200T/m/s). Patients with abandoned leads/fragments but without a generator in place do not require involvement by electrophysiology. Radiologist assessment for indication, risk vs benefit discussion with patient, and signed consent.
Pacemakers/ICDs
Newly implanted pacemakers and implanted cardioverter/defibrillators are considered MR conditional. However there are approximately 6 million patients with legacy devices, and >50% of these patients will have a clinical indication for MRI during the lifetime of the device.
Potential risks of MR imaging in patients with implanted cardiac devices include movement and/or vibration of the generator and leads, temporary or permanent malfunction of the device, and excessive heating or induced currents in the leads. However, a large body of evidence has demonstrated a low overall rate of complications. Currently there is broad international agreement that both MR conditional and non-MRI-conditional cardiac electronic implanted devices can be safety imaged, including statements from the following groups:
- Heart Rhythm Society
- American Heart Association
- American College of Radiology
- Canadian Heart Rhythm Society
- German Cardiac Society
- British Cardiovascular Society
Specific guidelines for patients with pacemakers/ICDs
The CEID/pacemaker/ICD in place for minimum of 6 weeks is strongly preferred.
Monitoring
Patient monitoring will be performed by a provider with advanced cardiac life support (ACLS) training and MRI safety training for all CEIDs/pacemakers/ICDs.
Continuous visual and voice contact must be maintained with the patient.
In the event that a patient is unresponsive or MRI is requested with anesthesia, radiology attending approval/assessment is required and the exam will only be performed in patients with MR conditional devices.
Exam scheduling
Patients should be scanned during normal workday hours (8am to 5pm) on select 1.5T scanners. 3T will only be considered for appropriate MRI conditional devices.
Device vetting
The device will enter the implant queue for identification and vetting. Verify that documentation of model, manufacturer, lead information is accurate and list most current system components, including revisions, generator replacement.
Expedited approval is sometimes possible if:
- an MRI conditional device is documented in the electronic medical record; and
- an established, modified MRI scan protocol appropriate for cardiac pacemakers/ICDs will be used.
To facilitate this, the specific device in question and MRI scan protocol should be clearly stated in the MRI requisition. Note that established scan protocols are in place for:
- Brain
- Cardiac
- Abdomen
- Knee
- Spine following the manufacturer's guidelines regarding MRI protocol restrictions.
MRI conditional versus non-MRI-conditional status
MRI conditional versus non-MRI-conditional status of the generator, leads, and combination thereof should be determined by UCSF Implant vetting team, principal/senior MR Technologist, MR Technologist Supervisor or MRI MD.
- If the entire CEID system is deemed MR conditional:
- The manufacturer MRI safety labelling will be followed including:
- Static field strength (1.5T vs. 3T)
- Maximum spatial field gradient
- Slew rate
- Specific absorption rate (SAR) limits (most commonly ≤2 W/kg)
- Anatomic location of isocenter
- Scan duration
- Coil restrictions
- Normal operating mode versus first-level controlled mode
Workflow for MRI conditional cardiac pacemakers/ICDs
- Prior to scheduling, device and leads identified and assessed for conditional/non-MRI-conditional status by any of these roles: UCSF Implant vetting team, principal/senior MR Technologist, MR Technologist Supervisor or MRI MD.
- No radiology MD approval is required if scanned per manufacturer recommendations.
- Device rep (remote or in person) will interrogate and reprogram device at the time of scan.
- Monitoring will be performed by a trained radiology nurse for both pacemaker dependent and nondependent patients.
- Device rep (remote or in person) verifies pacemaker function following completion of the exam.
If any issue arises (i.e., pain, discomfort, significant change in heart rate), the patient will be removed from the MRI scanner and the EP consult fellow will be called at 443-UCEP. If the patient is unresponsive or unstable, a code will be called. An external defibrillator should be easily accessible.
If the system is non-MRI-conditional, the following safety limitations will be implemented:
- MD radiologist approval for appropriateness to proceed
- Risk vs Benefit discussion with signed consent
- Minimum number of MRI sequences performed to answer the diagnostic question
- Depending on patient clinical scenario, low SAR protocols may be performed
- 1.5T static field strength
- Normal Operating Mode with SAR (≤1.5 or ≤ 2 W/kg) and dB/dt limitations (<200T/m/s)
- Coil limitations
- Body coil can be used for RF transmission
- Local transmit/receive coils if not positioned directly over the device
Workflow for non-MRI-conditional cardiac pacemakers/ICDs
- Prior to scheduling, device and leads identified and assessed for conditional/non-MRI-conditional status by any one of these roles: UCSF Implant vetting team, principal/senior MR Technologist, MR Technologist Supervisor or MRI MD.
- MD radiologist approval (QC fellow or section MRI safety representative) re: appropriateness to proceed – study indicated, presence of reasonable non-MRI alternative.
- Dependency/nondependency is evaluated per electrophysiology (EP) recommendations from pre-scan visit.
- Consent is obtained by radiology MD.
- EP Nurse Practitioner, EP Physician or EP Registered Nurse must interrogate and reprogram device at the time of scan.
- Monitoring for pacemaker dependent patients is performed by an EP nurse. Monitoring for pacemaker nondependent patient is performed by a trained radiology nurse.
- EP Nurse Practitioner, EP Registered Nurse or EP physician verifies pacemaker function following completion of the scan.
For non-MRI-conditional devices, risk-benefit analysis should clearly show the benefits of MRI. The study should not be performed if similar clinical information could be obtained with another image modality.
Assessment by EP (either inpatient consult or appointment with the EP clinic) should be schedule prior to the MRI date to review the details of the pacer/ICD and leads, and confirm that MRI is appropriate. EP should document in their notes regarding the encounter that a full discussion of risks and benefits of MRI was performed with the patient.
Device programming
Cardiac implanted electronic devices should be interrogated both before and after Cardiovascular Magnetic Resonance Imaging, with involvement of either electrophysiology providers or device representatives depending on the conditional/non-MRI-conditional status of the device. The device should be reprogrammed for the MRI including setting in either non-pacing or asynchronous pacing mode. Defibrillator therapies will be turned off, and magnet mode will be disabled. The device will be reprogrammed at the end of the exam.
Cardiac Loop Recorders
Cardiac Loop Recorders are MRI conditional devices. Cardiac Loop Recorders typically contain no lead wires or large loops of electrically conductive material. Product details regarding the specific loop recorder in question should be reviewed prior to MRI.
The main issue to consider with MRI is that data stored on the device may be altered or erased. Data should be downloaded before the time of study, and this issue should be discussed with the cardiologist who manages the loop recorder.
Because loop recorders contain ferromagnetic components, patients may feel slight movement during scanning. While not a safety hazard, patients should be made aware of this before scanning.
Summary and Key Points for On-Call Residents
General points:
- CT of the fetus should be avoided in all trimesters of pregnancy, because it may cause up to a doubling of the risk of fatal childhood cancer.
- No radiological procedure involving ionizing radiation to the pelvis should be undertaken in a patient who declares she may be pregnant without consultation with radiology faculty.
- MRI poses no known risk to the fetus in the second and third trimester. MRI in the first trimester should only be performed after consultation with radiology faculty.
- Breast feeding can be continued without interruption after administration of iodinated contrast or gadolinium to a lactating patient
- It is advisable to obtain written informed consent for CT of the abdomen or pelvis in a pregnant patient. For studies that pose minimal risk (including CT pelvimetry, CT of other body parts, and MRI) it is advisable to explain the negligible nature of the risk to the patient and document this discussion in either the chart or the radiology report.
Specific points:
- The most common indications for urgent CT during pregnancy are:
- Appendicitis - For first and second trimester pregnancies US and/or MR should be performed prior to obtaining a CT
- Pulmonary embolism - In this case a CT pulmonary angiogram exposes the fetus to less radiation than a VQ scan. Therefore, CT should be the initial modality.
- Renal colic - US is the initial study of choice.
- Trauma. US may be sufficient for the initial imaging evaluation of a pregnant patient who has sustained trauma, but CT should be performed if serious injury is suspected.
- All patients undergoing CT of the abdomen or pelvis during pregnancy should sign the written informed consent form available at (consent form). The consent form can be completed by either the referring physician or the involved radiologist (including the radiology resident on-call). Patients referred from the Department of Obstetrics, Gynecology and Reproductive Sciences will be consented by the referring physician.
- For studies that pose minimal risk (including CT pelvimetry, CT of other body parts, and MRI) it is advisable to explain the negligible nature of the risk to the patient and document this discussion in either the chart or the radiology report. This discussion can be undertaken by either the referring physician or the involved radiologist.
- CT contrast seems safe to use in pregnancy and should be administered in the usual fashion ñ this is far preferable to repeating a study because the initial examination was non-diagnostic due to lack of intravenous contrast.
- Intravenous gadolinium is contra-indicated in pregnancy, and should only be used if absolutely essential and only after discussion of risks and benefits with the patient and referring clinician and radiology faculty.
- Pelvimetry can be performed either by low dose CT or by MRI, and written informed consent is not required.
For more information, visit:
For more information, visit our Adult or Pediatric section:
| CT Scanners | ||
|---|---|---|
| Imaging Site | Maximum Weight | Maximum Diameter |
| Parnassus Hospital | 450 lbs | 70 cm |
| Parnassus PET/CT | 500 lbs | 78 cm |
| Medical Building 1 400 Parnassus (Outpatient) |
450 lbs | 70 cm |
| Mission Bay Hospital | 450 lbs | 80 cm |
| Precision Cancer Medicine Building (Outpatient) |
675 lbs | 70 cm |
| China Basin (Outpatient) | 450 lbs | 70 cm |
| Mount Zion Hospital | 450 lbs | 70 cm |
| Radiology at 2330 Post Street (Outpatient) |
450 lbs | 70 cm |
| Berkeley Outpatient Center | 500 lbs | 78 cm |
| MRI Scanners | ||
|---|---|---|
| Imaging Site | Maximum Weight | Maximum Diameter |
| Parnassus Hospital | 450 lbs | 70 cm |
| Medical Building 1 400 Parnassus (Outpatient) |
500 lbs | 70 cm |
| Mission Bay Hospital | 550 lbs | 70 cm |
| Precision Cancer Medicine Building (Outpatient) |
450 lbs | 70 cm |
| China Basin (Outpatient) | 450 lbs | 70cm (for limited access to 80cm - contact MR team) |
| Radiology at 2330 Post Street (Outpatient) |
550 lbs | 70 cm |
| Berkeley Outpatient Center | 550 lbs | 70 cm |
*Diameter is often the limiting factor for MRI. Maximum diameter listed does not account for padding and coils necessary for safe and effective imaging. Padding and coils may decrease the maximum patient diameter by up to 5 cm. Please see MRI section for more details.
| Interventional Radiology | |
|---|---|
| Imaging Site | Maximum Weight |
| Parnassus Hospital | 551 lbs |
| Mission Bay Hospital | 606 lbs |
| Precision Cancer Medicine Building (Outpatient) |
606 lbs |
| Mount Zion Hospital | 444 lbs |
For more information, visit:
Size and Weight Considerations for Imaging and Image-Guided Interventions
Connect with a Radiologist
Additional Resources

Radiology Locations
With locations in San Francisco and beyond, we offer convenient locations for your imaging care.

Radiology Services & Exams
We offer advanced imaging services and expert care to support diagnosis, treatment, and patient well-being.